ХРОМАТОГРАФІЯ (грец. chrоma — колір + grapho — пишу) у фармацевтичному аналізі — методи хроматографічного розділення речовин — це багатоступінчасті методи, в яких компоненти зразка розподіляються між двома фазами, одна з яких є нерухомою (стаціонарною), а інша — рухомою. Нерухома фаза може бути твердою або рідкою, нанесеною на твердий носій або гель. Нерухома фаза може бути упакована в колонку, нанесена як шар або як плівка. Рухома фаза може бути газом чи рідиною або флюїдом (газом у надкритичному стані). Розділення може ґрунтуватися на адсорбції, розподілі, іонному обміні, їх комбінації тощо, або на відмінностях у фізико-хімічних властивостях молекул (розмір, маса, об’єм тощо). Х. може використовуватися для отримання чистих сполук (препаративна Х.), кількісного та якісного визначення розділених компонентів (хроматографічний аналіз), а також для визначення фізико-хімічних характеристик розділених сполук. Хроматографічні системи класифікують за агрегатним станом фази (системи газ — рідина, газ — тверде тіло, рідина — тверде тіло, рідина — рідина), за геометрією хроматографічної системи (колонкова та планарні системи), за механізмом, що лежить в основі рівноважного розподілу між рухомою і нерухомою фазами (розподіл між двома рідинами — розподільна Х., адсорбція з рідини на поверхню твердого тіла — адсорбційна Х., іонний обмін — іонообмінна Х.) тощо. У фармацевтичному аналізі зазвичай класифікують такі види хроматографічних методів (планарні варіанти): Х. на папері (ХП), ТШХ; колонкові системи: газова Х., рідинна Х. (часто — ВЕРХ), ексклюзивна Х., флюїдна (надкритична) Х. Раніше поширеним був поділ Х. на адсорбційну і розподільну (газоадсорбційна, газорідинна, рідинна адсорбційна, рідинна розподільна), але згодом його визнали таким, що не повністю відображає структуру. Х. (як колонкова рідинна адсорбційна Х.) була відкрита М.С. Цвєтом у 1901 р. Однак бурхливий розвиток різних видів Х. розпочався у 30-ті роки ХХ ст.: 1938 р. — відкриття ТШХ, 1941 р. — відкриття колонкової рідинної розподільної Х., 1943 р. — відкриття розподільної паперової Х., 1952 р. — розроблення першого газового хроматографа.
ТШХ — планарний варіант рідинної Х., це метод розділення, у якому використовується нерухома фаза, що складається з відповідного матеріалу, нанесеного у вигляді стандартизованого тонкого шару і зафіксованого на основі (пластинці) зі скла, металу або пластмаси. Перед хроматографуванням розчини речовин, що аналізують, наносять на пластинку. Розділення засновано на процесах адсорбції, розподілу, іонного обміну та їх комбінації й здійснюється за допомогою переміщення в тонкому шарі (нерухомій фазі) досліджуваних речовин, розчинених у розчиннику або у відповідній суміші розчинників (рухомій фазі). Як сорбенти можуть використовуватися оксид алюмінію, целюлоза тощо, але у фармацевтичному аналізі найчастіше використовують стандартизований подрібнений силікагель з розміром часток 5–40 мкм (звичайні ТШХ-пластини) або 2–10 мкм (пластини для високоефективної ТШХ). Звичайно сорбент закріплюють на пластині зв’язувальною неорганічною (гіпс) або органічною (крохмаль) речовиною. Сорбент може бути силанізованим і містити флуоресцентний індикатор з інтенсивністю поглинання 254 (інколи 366) нм. Готові пластини можуть бути також імпрегновані (просочені) нерозчинною в рухомій фазі в’язкою рідиною. Хроматографічна камера — ємність з кришкою, що сильно прилягає, плоским дном або дном з двома жолобами з інертного прозорого матеріалу, що відповідають за розміром використовуваним пластинкам. Для горизонтального елюювання хроматографічна камера має жолоб для рухомої фази і додатково містить пристрій для подавання рухомої фази до нерухомої. Найчастіше використовують метод вертикального (висхідного) елюювання. Стінки хроматографічної камери вистилають фільтрувальним папером. Рухому фазу наливають у камеру в кількості, достатній для того, щоб після змочування фільтрувального паперу покрити дно камери шаром рідини, необхідним для хроматографування. Для насичення хроматографічну камеру з рухомою фазою закривають кришкою й витримують протягом 1 год при температурі від 20 до 25 °С. Об’єми розчинів аналізованих речовин наносять невеликими порціями, одержуючи смуги або круглі плями на підхожій відстані від нижнього краю та від бічних країв пластинки. Розчини наносять на лінію, паралельну нижньому краю пластинки, на відстані не менше 10 мм між пробами. Після випаровування розчинників з нанесених проб пластинку поміщають у хроматографічну камеру якомога більш вертикально, стежачи за тим, щоб плями або смуги знаходилися вище поверхні рухомої фази. Камеру закривають, залишають її при температурі від 20 до 25 °С у захищеному від прямих сонячних променів місці. Після того як рухома фаза пройде необхідну відстань, зазначену в окремій статті, пластинку виймають, сушать і виявляють плями зазначеним способом. У випадку двовимірної Х. після першого хроматографування пластинку сушать і роблять друге хроматографування у напрямку, перпендикулярному до першого. Для виявлення (проявлення плям) речовин на пластинках використовують УФ і видиме світло, флуоресценцію, пари йоду, різні хімічні проявники, якими обприскують пластину. Для кількісного визначення використовують різні детектори (спектрофотометричні, радіоактивні тощо). У цьому випадку зазвичай використовують горизонтальне елюювання. Однак ці методи використовують досить рідко, оскільки вони поступаються рідинній колонковій Х. Основне застосування ТШХ у фармацевтичному аналізі — ідентифікація й граничний контроль домішок. Метод як простий і ефективний найчастіше використовують у невеликих лабораторіях. У фармакопейних монографіях його поступово заміняють на більш універсальну і точну ВЕРХ.
Процедура ХП аналогічна ТШХ, лише тонкий шар замінюється на папір. Характерними ознаками ХП є широке використання низхідного елюювання і порівняно з ТШХ досить тривалий час хроматографування (кілька годин). Основне застосування ХП у фармацевтичному аналізі — ідентифікація й граничний контроль домішок. Останнім часом використовується рідко, оскільки замінюється на більш універсальну ТШХ.
Газова Х. — це метод розділення, в якому рухомою фазою є газ (газ-носій), а нерухомою — тверда речовина або рідина, яка нанесена на твердий інертний носій або рівномірно покриває внутрішні стінки колонки (нерухома фаза поміщена у колонку). Газова Х. заснована на механізмах адсорбції і/або розподілу. Обладнання складається з системи подавання газу, пристрою для введення проби, хроматографічної колонки, детектора і реєструючого пристрою. Колонки зазвичай виготовляють зі скла або нержавіючої сталі й заповнюють нерухомою фазою. Газ-носій проходить із заданою швидкістю через пристрій уведення проби, колонку, а потім — через детектор. Визначення проводять при сталій температурі або відповідно до заданої температурної програми. Детектори газової Х. (полуменево-іонізаційний, за теплопровідністю, термоіонний, мас-спектрометричний, за захопленням електронів тощо) найбільш універсальні й чутливі серед інших хроматографічних методів. Колонку, пристрій уведення проби і детектор термостатують при зазначеній температурі. Готують розчин, який випробовують, і розчин (-и) порівняння, як зазначено в окремій статті. Використовуючи розчини порівняння, налагоджують прилад і добирають об’єми проб, що вводяться і дозволяють одержати необхідний сигнал. Виконують повторні введення для перевірки збігу сигналу і перевіряють, якщо необхідно, кількість теоретичних тарілок. Уводять розчини і реєструють результати хроматографування. Визначають висоту (при ізотермічному визначенні, якщо коефіцієнт симетрії становить 0,8–1,2) або площу піків аналізованих компонентів. Для розрахунків вмісту аналізованого компонента використовують методи зовнішнього або внутрішнього стандарту. Для розрахунку вмісту домішок може використовуватися, де це можливо, метод внутрішньої нормалізації. У цих випадках рекомендують застосовувати широкодіапазонний підсилювач та автоматичний інтегратор. У фармацевтичному аналізі газову Х. використовують для контролю залишкових кількостей органічних розчинників у субстанціях (обов’язковий фармакопейний метод), ідентифікації й кількісного визначення розчинників та інших летких сполук (напр. етерних олій), а також ЛП.
Рідинна Х. — це метод розділення, у якому рухомою фазою є рідина, а нерухомою фазою, поміщеною в колонку, — тонкодисперсна тверда речовина або рідина, нанесена на твердий тонкодисперсний носій, у т.ч. хімічно модифікований шляхом уведення органічних груп. Рідинна Х. заснована на механізмах адсорбції, розподілу, іонного обміну або розподілу за розмірами молекул. Залежно від природи рухомої фази в адсорбційній рідинній Х. розрізняють нормально-фазову та оборотно-фазову Х. У нормально-фазовій Х. нерухома фаза — полярна (найчастіше силікагель), а рухома фаза — неполярна (гексан або суміш гексану з більш полярними органічними розчинниками — хлороформом, алкоголями тощо). Затримка речовин зростає зі збільшенням їх полярності. У нормально-фазовій Х. елюювальна здатність рухомої фази збільшується зі зростанням її полярності. У зворотно-фазовій Х. нерухома фаза — неполярна (гідрофобні силікагелі з привитими групами С8, С18); рухома фаза — полярна (суміш води і полярних розчинників; ацетонітрилу, метанолу, тетрагідрофурану тощо). Затримка речовин збільшується з підвищенням їх гідрофобності (неполярності). Найменшу елюювальну здатність має вода, а для підвищення елюювальної здатності в рухому фазу вводять ацетонітрил, метанол та інші розчинники. Обладнання зазвичай складається із системи подавання рухомої фази, пристрою введення проби (з використанням шприца або петльового дозатора), хроматографічної колонки, детектора і реєструючого пристрою. Рухома фаза зазвичай подається під тиском з однієї або кількох посудин і протікає через пристрій уведення проби, колонку, а потім — через детектор із заданою швидкістю. Температуру хроматографічної колонки підтримують сталою. Склад рухомої фази залежно від зазначеного в окремій статті може або залишатися сталим протягом усього аналізу (ізократичне елюювання), або змінюватися відповідно до заданої програми (градієнтне елюювання).
Детектування. У фармацевтичному аналізі зазвичай використовують такі універсальні детектори: спектрофотометричний, рефрактометричний, електрохімічний, а також мас-спектрометричний, флуориметричний тощо. Методика. Колонку врівноважують при зазначеному складі рухомої фази. Готують розчин, який аналізують, і розчин(-и) порівняння. Розчини не мають містити твердих часток. Використовуючи розчини порівняння, налагоджують прилад і підбирають об’єми проб, що вводяться, які дозволяють одержати необхідний сигнал. Уводять розчини і реєструють результати хроматографування. Визначають висоту (при ізократичному елююванні, якщо коефіцієнт симетрії становить 0,8–1,2) або площу піків аналізованих компонентів. Для розрахунків вмісту аналізованого компонента зазвичай використовують метод зовнішнього та іноді внутрішнього стандартів. Для розрахунку вмісту домішок може використовуватися, де це можливо, метод внутрішньої нормалізації. У цих випадках рекомендують використовувати широкодіапазонний підсилювач та автоматичний інтегратор. У фармацевтичному аналізі рідинну Х. використовують як один з найефективніших і поширених методів ідентифікації, контролю домішок і кількісного визначення ЛП та контролю їх виробництва. Застосування рідинної Х. для контролю чистоти субстанцій стає обов’язковим.
Ексклюзивна Х. — окремий вид рідинної колонкової Х., хроматографічний метод, в якому процес розподілу молекул речовин у розчині відбувається відповідно до їх розмірів. При використанні органічної рухомої фази метод називають гель-проникаючою Х., а при використанні водної рухомої фази — гель-фільтраційною Х. Проба вводиться в колонку, заповнену гелем або пористими частками наповнювача, і переноситься рухомою фазою через колонку. Розподіл за розмірами відбувається за рахунок багаторазового обміну молекул розчиненої речовини між розчинником рухомої фази і цим самим розчинником (нерухома фаза) у порах матеріалу, яким заповнена колонка. Діапазон розмірів молекул, що розділяють, визначається діапазоном розмірів пор наповнювача. Досить маленькі молекули, здатні проникати в усі пори матеріалу стовпчика, елююються в повному об’ємі колонки (Vt — повний проникаючий об’єм або межа ексклюзії). Молекули речовини, що перевищують за розміром усі пори матеріалу колонки, не стримувані ним, мігрують лише через простір між частками наповнювача і елююються у вільному об’ємі колонки (V0 — об’єм ексклюзії, або мертвий об’єм). Розподіл молекул речовин за розмірами відбувається між вільним об’ємом і повним об’ємом колонки; найбільш ефективний розподіл відбувається в перших двох третинах цього діапазону. Специфічною частиною обладнання є хроматографічна колонка відповідних розмірів, заповнена матеріалом, що забезпечує розподіл молекул за розмірами у потрібному діапазоні. Якщо необхідно, колонку термостатують. Через колонку з постійною швидкістю пропускають елюент. До одного кінця колонки приєднують пристрій уведення проби, напр. інжектор з припиненням потоку, шприцевий інжектор з мембраною для введення проби без припинення потоку або петльовий інжектор з клапаном, що перемикає потік. До цього кінця колонки також може бути приєднаний відповідний насос для подавання елюенту з контрольованою швидкістю. Проба може також наноситися безпосередньо на суху поверхню матеріалу колонки або, якщо густина проби перевищує густину елюенту, нашаровуватися на поверхню матеріалу колонки під елюент. Інший кінець колонки приєднують до відповідного детектора з пристроєм, що реєструє й забезпечує контроль відносних концентрацій розподілюваних компонентів проби. Як наповнювач може використовуватися або м’який матеріал (набухлий гель), або жорсткий (пористе скло, силікагель), або поперечнозшитий органічний полімер, який підходить для цього розчинника. При використанні жорстких матеріалів застосовують подавання рухомої фази під тиском, що прискорює розподіл. Рухому фазу вибирають з урахуванням природи проби, наповнювача і методу детектування. Зазвичай використовують такі детектори: фотометричний, рефрактометричний або люмінесцентний. Якщо необхідно, може бути приєднаний автоматичний колектор фракцій. Основне застосування у фармацевтичному аналізі — аналіз полімерів (напр. декстранів).
Флюїдна (надкритична) Х. — метод хроматографічного розділення, в якому рухома фаза є флюїдом (газом у надкритичному або субкритичному стані). Нерухома фаза, поміщена в колонку, є добре поділеними твердими частками (силікагель або пористий графіт), хімічно модифікованою нерухомою фазою (використовують у рідинній Х.) або поперечно зв’язаною рідкою плівкою, рівномірно нанесеною на стінки колонки для капілярних колонок. Флюїдна Х. ґрунтується на механізмах адсорбції або масового розподілу. Обладнання складається з насосної системи, що охолоджується, інжектора, хроматографічної колонки, поміщеної в термостат, детектора, регулятора тиску і приладу для оброблення даних (інтегратор або самописець). Рухому фазу становить оксид вуглецю, який може містити полярні модифікатори (метанол, 2-пропанол і ацетонітрил). Склад, тиск (густина), температура та швидкість її потоку можуть бути постійними або змінюватися згідно із заданою програмою. Звичайно використовують спектрофотометричний або полуменево-іонізаційний детектори. Можуть також використовувати інші спеціальні детектори. Флюїдна Х. поєднує в собі переваги рідинної Х. (висока розчинювальна здатність) і газової Х. (висока ефективність та універсальне детектування). Її застосовують для важколетких і термолабільних сполук, особливо природного походження.
Основні величини і співвідношення у хроматографічному аналізі
Зв’язок об’єму (VR) і часу (tR) утримання:
VR = tR · v,
Величина Rf (планарні варіанти — ТШХ, ХП):
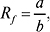
де a — відстань середини плями після хроматографування від точки нанесення плями («старту»); b — відстань фронту рухомої фази від «старту». Для «хвостатих» плям середина плями є невизначеною, для них (з метою ідентифікації) величину а інколи вимірюють від верхнього краю після хроматографування. Зв’язок колонкової Х. (напр. ВЕРХ) з планарними варіантами (ТШХ):
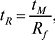
де tM (часто позначається також як t0) — мертвий час колонки (відповідає компоненту, що не утримується). Замість часу утримування нерідко використовують об’єм утримування. Це рівняння приблизне, оскільки ВЕРХ на відміну від ТШХ, працює в режимі насичення сорбенту. Виправлений час утримування:
t’R = tR – tM.
Коефіцієнт масового розподілу (Dm) (часто називають коефіцієнтом ємності k‘) і величина Rm:
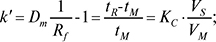 Rm=lgk‘,
Rm=lgk‘,
де KC — константа розподілу, VS — об’єм стаціонарної фази, VM — об’єм рухомої фази. Коефіцієнт масового розподілу Dm дорівнює відношенню кількості речовини, що хроматографується, в стаціонарній і рухомій фазах. Коефіцієнт симетрії піку:
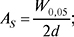
де W0,05 — ширина піку на одній двадцятій частині його висоти; d — відстань між перпендикуляром, опущеним з максимуму піку, і передньою межею піку на одній двадцятій його висоти.
Характеристики розділення двох речовин. Планарні варіанти:
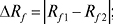
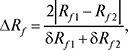
де δRf1 і δRf2 — діаметри плям у величинах Rf. Величина Rs (коефіцієнт розділення) мало застосовується при невеликих значеннях Rf, оскільки її максимум нерідко досягається при значеннях Rf, близьких до нуля. Колонкові варіанти:
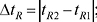
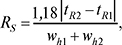
де wh1 і wh2 — ширина піку (у тих же одиницях, що й tR) на половині висоти. Rs = 1 відповідає 98% розділення, Rs = 1,5–99,7% (до базової лінії). У планарних варіантах (ТШХ і ХП) переважно використовують величини ∆Rf, у колонкових (ВЕРХ) — коефіцієнт розділення Rs.
Ефективність пластинки або колонки (число теоретичних тарілок). Планарні варіанти:
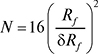 .
.
Колонкові варіанти:
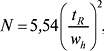
де wh — ширина піку на половині висоти в тих же одиницях, що й tR. Ця величина не застосовується при tR, близькому до tM. Тому іноді використовують величину, яка не має цього дефекту:
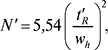
Відношення сигнал/шум (S/N) розраховують за формулою:
S/N = 2H/hn,
де H — висота піку відповідного компонента на Х.; hn — абсолютне значення найбільшої флуктуації шуму базової лінії на хроматограмі холостого розчину, яке спостерігається на проміжку, що дорівнює двадцятикратній ширині на напіввисоті піку на хроматограмі розчину порівняння, розміщеному рівномірно навколо місця розташування піку.
Зв’язок утримування зі складом рухомої фази
На практиці для бінарної рухомої фази рідинної Х. (ТШХ, ХП, ВЕРХ) найчастіше використовують рівняння Сочевинського:
Rm = a – b•lg C2,
де а і b — деякі емпіричні параметри, С — молярна або об’ємна частка активного компонента.
Рівняння застосовують тільки для бінарних рухомих фаз без екстремумів утримування з великими відмінностями в елюювальних силах компонентів. Дає Rf = 0 для хроматографування в чистому першому (неактивному) компоненті рухомих фаз (С2 = 0). Узагальнене рівняння Сочевинського використовують для багатокомпонентної рухомої фази (модель єдиного адсорбційного центру):
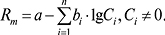
Підсумовування здійснюється за всіма n по всіх n‘-компонентах рухомої фази; bi — деякі емпіричні параметри. Це рівняння застосовують для будь-якої кількості компонентів рухомої фази за наявності й відсутності екстремумів утримування. Співвідношення величин bi характеризує відносну елюювальну силу компонентів рухомої фази на даному сорбенті в даній n-компонентній рухомій фазі для даної сполуки. Для багатокомпонентної рухомої фази використовують також рівняння Снайдера – Яронеця:
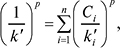
де р — емпіричний «параметр гетерогенності», а також рівняння Осцика:
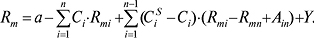
При обчисленнях звичайно враховують:
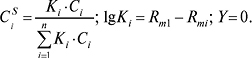
Відносні залежності
Відносні величини утримування.
Колонкові варіанти — відносний час (об’єм) утримування:
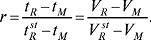
Планарні варіанти:
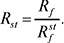
Індекс st відносять до речовини, яку визнано стандартом. Ці величини менше залежать від умов експерименту, ніж самі абсолютні величини, і тому їх широко застосовують при ідентифікації. Для бінарних рухомих фаз властиве співвідношення:
Rm = a•Rmst + b,
де а, b — емпіричні параметри. У трикомпонентних рухомих фазах ця пряма переходить у відповідний трикутник.
Рівняння Перішо зв’язує значення Rm однієї й тієї ж речовини в багатокомпонентних рухомих фазах Х і Y:
Rm (Y) = a•Rm (X) + b,
де а, b — емпіричні параметри.
Функція стійкості бінарної рухомої фази.
Планарні варіанти:
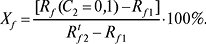
Колонкові варіанти:
Yk = C2(k’ = 1) + C2(k’ = 10).
Функції стійкості рухомої фази характеризують швидкість зміни величин утримування під впливом зміни складу бінарної рухомої фази. При ТШХ величини Xf характеризують частку загальної зміни значень Rf на відрізку C2 = 0 — 0,1 активного компонента. Бажано, щоб величина Хf була меншою 10%. При ВЕРХ величина Yk характеризує інтервал молярної або об’ємної частки активного компонента, що розміщується на найбільш практично важливій ділянці значень k‘=1–10. Бажано, щоб величина Yk була в інтервалі 0,5–0,7.
Перевірка придатності хроматографічної системи
На хроматографічний процес впливає багато факторів, які досить складно піддаються стандартизації й суттєво впливають на невідтворюваність хроматографічних методик аналізу. Одним з найважливіших чинників є активність сорбенту. Тому в ДФУ висунуто досить суворі вимоги до стандартності сорбенту і готових ТШХ-пластинок, які використовують для аналізу. Однак це не може гарантувати необхідної відтворюваності методик контролю якості ЛП, які містять сполуки різної полярності. Крім того, у разі колонкових варіантів (особливо при використанні сорбенту з прищепленими групами) слід зважати на зміну характеристик сорбенту, пов’язану, напр., з «облисінням» прищеплених груп. Тому у Фармакопею США у 1990 р. уведено для ВЕРХ і газової Х. систему тестів — «Перевірка придатності хроматографічної системи». Це випробування, яке виконують перед проведенням власне аналізу і для якого звичайно використовують особливий «розчин для перевірки придатності», стало обов’язковим для всіх хроматографічних методів і було введено іншими фармакопеями, зокрема ДФУ. Формулювання цього випробування для планарних і колонкових варіантів відрізняється. Для планарних варіантів (ХТШ, ХП) за ДФУ хроматографічну систему зазвичай вважають придатною, якщо: на хроматограмі розчину порівняння, який використовується для перевірки придатності хроматографічної системи, чітко діляться плями зазначених в окремій ФС речовин; Rf основної плями на хроматограмі розчину, який випробовується, близький до величини, зазначеної в окремій ФС; на хроматограмі розчину порівняння, який використовується для перевірки чутливості хроматографічної системи, чітко видно пляму. Для колонкових варіантів (ВЕРХ, газова Х.) хроматографічну систему зазвичай вважають придатною, якщо: відносний час утримування зазначених речовин близький до вказаних значень; кількість теоретичних тарілок (ефективність хроматографічної системи), розрахована за зазначеним піком, не менша від зазначеної величини; коефіцієнт розділення зазначених піків, розрахований з хроматограм розчину порівняння, не менший від зазначеної величини; відносне стандартне відхилення, розраховане для висоти або площі зазначеного піку або їх відношень до висоти чи площі, або висоти піку внутрішнього стандарту з хроматограм розчину порівняння, не більші від зазначеної величини; коефіцієнт симетрії зазначеного піку, розрахований з хроматограм розчину порівняння, перебуває в зазначених межах. Останнім часом дві останні вимоги виносять у загальну ФС і тому не включають до тесту на придатність хроматографічної системи. Для забезпечення виконання вимог тесту «Перевірка придатності хроматографічної системи» допускають модифікацію хроматографічних умов, описаних в окремій ФС.
Георгиевский В.П., Гризодуб А.И. Стандартизация и контроль качества лекарственных средств // Технология и стандартизация лекарств. — Х., 1996; Гризодуб А.И., Георгиевский В.П. Линейные зависимости в тонкослойной хроматографии с двухкомпонентными системами растворителей // Журн. аналит. химии. — 1981. — Т. 36, № 5; Гризодуб А.И., Георгиевский В.П. Устойчивость значений Rf и Rm к колебаниям в составе бинарных подвижных фаз // Журн. физ. химии. — 1984. — Т. 58, № 6; ДФУ. — Х., 2001. Доповнення 1. — Х., 2004; Осцик Я. Некоторые проблемы термодинамической адсорбции из многокомпонентных растворов // Физическая адсорбция из многокомпонентных фаз. — М., 1972; European Pharmacopoeia. Chromatographic separation techniques, 2004; Jaroniec M., Martire D.E., Borowko M. Theoretical basis of liquid adsorption chromatography with mixed mobile phases and its connection with theory of adsorption // Interface Sci. — 1985. — Vol. 22, № 2–4; Model of Mixed Adsorpion Centre for Description of Retention in Liquid Chromatography with Multicomponent Mobile Phases / M.G. Levin, A.I. Grizodoub, N.N. Asmolova et al. // Chromatographia. — 1993. — Vol. 37, № 9/10; Perisho C.R. A two-parameters formula relating Rf values in different solvent systems // Anal. Chem. — 1968. — Vol. 40, № 3; Soczewinski E., Golkiewich W. Application of the law of mass action to thin-layer chromatography systems of the type electrone donor solvent-silica gel // Chromatographia. — 1971. — Vol. 4, № 11.