ВАЛІДАЦІЯ АНАЛІТИЧНИХ МЕТОДИК І ВИПРОБУВАНЬ. В.а.м. — це експериментальний доказ того, що методика придатна для вирішення передбачуваних завдань. Випробування — це аналітична методика, описана в окремій статті, в сукупності з вимогами до одержуваних за нею результатів. Результатом проведення випробувань є відповідь на питання, чи відповідає цей ЛП вимогам окремої статті. При контролі якості ЛП або контролі їх виробництва зазвичай мають справу з аналітичними випробуваннями, вимоги до результатів яких визначають критерії В. При проведенні В.а.в. ЛП передбачається, що аналітичне обладнання відповідає вимогам ДФУ. У разі, якщо цих вимог недостатньо, необхідна відповідна кваліфікація обладнання з уведенням додаткових вимог до АНД. В.а.м. проводять лише на атестованому та перевіреному обладнанні.
Усі аналітичні методики і випробування, які входять до монографії чи АНД, мають бути валідовані. В.а.м. підлягають: випробування на ідентифікацію; кількісні випробування для визначення домішок; випробування на граничний вміст домішок; кількісні випробування для визначення діючої речовини та інших компонентів (напр. консервантів) у лікарських субстанціях і готових ЛП; зокрема, сюди входять «Кількісне визначення», «Однорідність вмісту діючої речовини в одиниці дозованого ЛП» і «Розчинення». Методики аналізу і випробування, які вже включені в окремі статті ДФУ, вважаються валідованими. У разі, якщо до АНД включається методика, яка не описана у ДФУ, необхідне проведення валідації у повному обсязі. Якщо методика описана у ДФУ, то вимоги до її валідації при включенні до АНД визначають по-різному. Для субстанцій: за наявності у виробника субстанції сертифіката відповідності монографії Європейської фармакопеї (ЄФ) валідація не потрібна. Якщо такий сертифікат відсутній, необхідно продемонструвати, що всі домішки (супутні домішки і залишкові кількості органічних розчинників) контролюються монографією ЄФ. Якщо це не так, то слід ввести додаткові випробування з відповідною В. Для готових ЛП В.а.м. проводять в обмеженому обсязі, в її результаті необхідно продемонструвати, що для даного складу препарату і упаковки методики АНД дають коректні результати. Замість фармакопейних методик можуть використовувати альтернативні методики за умови, що вони дають еквівалентні результати. Для таких методик необхідна В.а.м. у повному обсязі. Обсяг валідаційних досліджень залежить від специфіки методу аналізу і конкретної методики. Напр., методика визначення сульфатної золи В.а.м. зазвичай не потребує. Водночас хроматографічні інструментальні методики потребують досить великого обсягу валідаційних досліджень. Якщо для фармакопейної методики немає необхідності у В.а.м., потрібно провести лише верифікацію методики, тобто експериментальне підтвердження того, що методика може бути коректно відтворена в лабораторному оточенні. При змінах у методиці може бути необхідною ревалідація (В.а.м. в обмеженому обсязі).
Основні поняття. Специфічність — здатність однозначно оцінювати аналізовану речовину в присутності інших компонентів, які можуть бути у зразку. Це можуть бути домішки, продукти розкладу, допоміжні речовини тощо. Недолік специфічності випробування може бути компенсований іншим (іншими) додатковим випробуванням. Специфічність для різних типів випробувань означає: ідентифікацію — доказ того, що ідентифіковано саме аналізовану речовину. Випробування на домішки — доказ того, що кожне випробування на домішки дозволяє однозначно характеризувати вміст домішок у зразку (напр. випробування «Супровідні домішки», «Важкі метали», «Вміст залишкових кількостей органічних розчинників» та ін.). Кількісне визначення (вміст або активність) — доказ того, що методика дозволяє точно і правильно встановити вміст або активність саме аналізованої речовини у зразку. Правильність характеризує ступінь відповідності між відомим справжнім значенням або довідковою величиною і значенням, одержаним за цією методикою. Точність аналітичної методики виражає ступінь близькості (або ступінь розкиду) результатів для серії вимірів, виконаних за даною методикою на різних пробах одного й того ж однорідного зразка. Вона може розглядатися на трьох рівнях: збіжність, внутрішньолабораторна точність і відтворюваність. Точність необхідно вивчати на вірогідно однорідних зразках. Однак якщо однорідний зразок одержати неможливо, можна використовувати його розчин або модельні суміші. Точність аналітичної методики зазвичай характеризують відхиленням, стандартним відхиленням або відносним стандартним відхиленням для серії вимірювань. Збіжність характеризує точність методики при її виконанні в одних і тих самих умовах (зокрема одним і тим самим аналітиком або групою аналітиків) протягом невеликого періоду. Вивчають, виконуючи не менше 9 визначень, що охоплюють діапазон застосування методик, або не менше 6 визначень для зразків із вмістом аналізованої речовини, близьким до номінального. Внутрішньолабораторна точність характеризує вплив внутрішньолабораторних варіацій: різні дні, різні аналітики, різне обладнання і подібні зміни. Відтворюваність характеризує точність у міжлабораторному експерименті. Оскільки на стадії розробки методики зазвичай складно провести міжлабораторні дослідження, для характеристики відтворюваності можуть застосовувати прогноз невизначеності методики аналізу, який ґрунтується на фармакопейних вимогах (або вимогах АНД) до обладнання й аналітичного посуду. У деяких випадках, напр. у багатокомпонентному спектрофотометричному аналізі, прогноз невизначеності аналітичної методики є обов’язковим. Межа виявлення (МВ) для конкретної аналітичної методики — мінімальна кількість аналізованої речовини у зразку, яка може бути виявлена (при цьому не обов’язково має бути визначене точне значення). Для встановлення межі виявлення використовують візуальну оцінку, співвідношення сигнал/шум (2:3), параметри калібрувальної прямої (стандартне відхилення вільного члена Sa і тангенс нахилу B) та ін. Останній підхід є більш об’єктивним:
МВ = 3,3 · Sa /В.
Межа кількісного визначення (МКВ) для аналітичної методики — мінімальна кількість аналізованої речовини у зразку, яка може бути кількісно визначена з необхідною правильністю і точністю. Межа кількісного визначення є валідаційною характеристикою методик кількісного визначення низьких концентрацій речовин у зразку і розглядається в основному при визначенні домішок і/або продуктів розкладу. Для визначення МКВ застосовують ті ж підходи, що й для МВ. Більш об’єктивним можна вважати підхід, який ґрунтується на параметрах лінійної залежності:
МКВ = 10 · Sa /В.
Лінійність — здатність методики (у межах діапазону застосування) давати величини, прямо пропорційні концентрації (кількості) аналізованої речовини у зразку. Діапазоном застосування аналітичної методики є інтервал між мінімальною і максимальною концентрацією (кількістю) аналізованої речовини у зразку (включаючи цю концентрацію), для якого показано, що аналітична методика має потрібну точність, правильність і лінійність. Мінімальні допустимі діапазони застосування методик: для кількісного визначення лікарських субстанцій або ЛП — 80–120% номінального вмісту; для однорідності дозування — 70–130% номінального вмісту, якщо для випробування не потрібний більш широкий інтервал (напр. для дозованих аерозолів); для випробувань на розчинення ±20% (абсолютний) нормованої величини вивільнення; для визначення домішок — від концентрації, у якій домішку зазвичай виявляють, до 120% нормованого вмісту. Робастність — здатність аналітичної методики не зазнавати впливу малих заданих (контрольованих) аналітиком змін в умовах виконання методики. Зокрема, вивчають стійкість у часі аналітичних розчинів, час екстракції тощо. Зазвичай на результати можуть впливати: різниця в досвіді аналітиків; умови навколишнього середовища (температура, вологість); реактиви (різні постачальники). У разі використання кількісної рідинної хроматографії вивчають вплив рН рухомої фази; складу рухомої фази; колонок (різні серії та/чи постачальники); температури; швидкості рухомої фази. У разі газової хроматографії вивчають вплив колонок (різні серії та/чи постачальники); температури; швидкості газу-носія. Робастність є показником надійності методики при її використанні у зазначених умовах. Валідаційні характеристики і вимоги. Набір досліджуваних характеристик аналітичних методик (випробувань), які підлягають В. (таблиця), залежить від призначення аналітичної методики. Типові валідаційні характеристики: правильність, точність, збіжність, внутрішньолабораторна точність, специфічність, межа виявлення, межа кількісного визначення, лінійність, діапазон застосування. Цей список слід розглядати як типовий для зазначених випробувань (аналітичних методик). Як правило, на стадії розробки методики вивчають також валідаційну характеристику — робастність.
Повторне проведення В.а.м. може бути необхідне у такому разі: зміна у синтезі лікарської субстанції; зміна у складі ЛП; зміни в аналітичній методиці.
Таблиця. Валідаційні характеристики, які розглядаються для різних випробувань і методик
| Характеристики | Типи аналітичних методик | |||
| Ідентифікація | Випробування на домішки | Кількісне визначення розчинення, визначення лише вмісту, активності |
||
| кількісні | граничні | |||
| Правильність | – | + | – | + |
| Точність: Збіжність Внутрішньолабораторна точність |
+ +* |
– – |
+ +* |
|
| Специфічність** | + | + | + | + |
| Межа виявлення | – | –*** | + | – |
| Межа кількісного визначення | – | + | – | – |
| Лінійність | – | + | – | + |
| Діапазон застосування | – | + | – | + |
Примітка: «–» — характеристика, що зазвичай не досліджується; «+» — характеристика, що зазвичай досліджується; *у тих випадках, коли проводять дослідження відтворюваності, дослідження внутрішньолабораторної точності не вимагається; **недолік специфічності випробування можна компенсувати іншим (іншими) додатковим випробуванням; ***може бути потрібним у деяких випадках (напр., коли межа визначення і нормована межа вмісту визначуваної домішки близькі).
Необхідною умовою проведення В.а.м. є наявність критеріїв прийнятності отриманих валідаційних характеристик. Напр., при вивченні збіжності методики кількісного визначення готового ЛП з допусками вмісту діючої речовини 95–105% номінального значення був отриманий 95% довірчий інтервал 2,5%. Чи є отримана збіжність достатньою для цієї методики кількісного визначення? Така сама ситуація виникає при вивченні всіх валідаційних характеристик. Тому без наявності науково обґрунтованих критеріїв неможливе проведення В.а.м. Критерії прийнятності методик пов’язані з допусками вмісту речовини, яка аналізується (або характеристики, що вивчається). При розробці критеріїв можна використовувати різні підходи, напр. підходи, які ґрунтуються на принципі незначущості невизначеності методики аналізу (або її складової) порівняно з допусками вмісту речовини, що аналізується. Враховуючи це, ДФУ для методик кількісного визначення рекомендує, щоб повна відносна невизначеність результату аналізу (∆Аs) у відсотках, виражена як однобічний відносний довірчий інтервал для рівня надійної ймовірності 95%, не перевищувала такі значення:
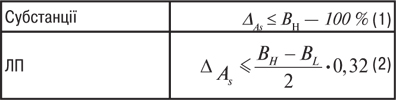
де BH — верхня межа вмісту за специфікацією у відсотках; BL — нижня межа вмісту за специфікацією у відсотках.
При дослідженні лінійної залежності зручно працювати в нормалізованих координатах, подаючи концентрації та аналітичний сигнал у відсотках до номінальних значень. У цьому разі застосування принципу незначущості й рівнянь (1–2) дозволяє сформулювати критерії прийнятності для параметрів лінійної залежності (остаточне стандартне відхилення розрахованої прямої RSDo, коефіцієнт кореляції Rc, вільний член а):

де RSDy — відносне стандартне відхилення для концентрацій, які використовувалися при вивченні лінійності; Сmin і Сst — мінімальна і номінальна концентрація діапазону застосування відповідно; Sst — номінальне значення аналітичного сигналу.
Аналогічно можна отримати вимоги до правильності, внутрішньолабораторної та міжлабораторної точності.
Документи з В.а.м. В.а.м. — невід’ємна частина Належної виробничої практики (GMP). Усю роботу з В.а.м. слід проводити структурованим шляхом відповідності з документованими процедурами для демонстрації того, що поставлених завдань було досягнуто. Найважливішим аспектом проведення В.а.м. згідно з GMP є належна система документації. При вивченні В.а.м. зазвичай оформляють такі документи: план проведення В., протокол В., звіт про відхилення (якщо потрібно), звіт з В., протокол передачі методики (якщо В. проводять не в тій лабораторії, в якій буде виконуватися аналіз). Проведення В.а.м. має бути заплановане у валідаційному майстер-плані. Отримані експериментальні дані документують згідно з протоколом В.а.м. і порівнюють з установленими критеріями. Первинні дані мають бути відповідним чином документовані й доступні за вимогою, оскільки вони можуть бути необхідні при інспектуванні. Протокол В.а.м. — письмовий план, в якому встановлено, як проходитиме В., включаючи валідаційні характеристики, критерії, устаткування і точки прийняття рішення про прийнятність одержаних результатів. Завершенням В.а.м. є оформлення письмового звіту про В. із зазначенням дати проведення В.а.м., обробки даних, аналізу і висновків за одержаними результатами у порівнянні з критеріями (результати мають відповідати критеріям і поставленому завданню). У разі потреби може бути проведене додаткове дослідження. Після отримання позитивних результатів звіт про В.а.м. затверджують, зазначивши дату із підписом. Звіт включає заголовок, мету проведення В., посилання на протокол В., подробиці про матеріали, що використовуються, опис аналітичного устаткування, програм і циклів (якщо застосовані), деталі аналітичної методики (умови виконання аналізу, налагодження приладів, попередження, інформація про реактиви і стандартні зразки, верифікація — перевірка придатності аналітичної системи), розрахункові формули і статистичні процедури. На підставі цього звіту може бути підготовлена скорочена версія для супроводу реєстраційного досьє. Проведення В. потребує значних затрат часу, коштів, залучення різних спеціалістів. Загальний обсяг валідаційних документів за АНД (до яких обов’язково включаються первинні дані) може сягати декількох сот сторінок.
Відповідальні особи. Організація проведення В.а.м. може бути здійснена різними способами, напр. за такою схемою. На підприємстві призначають валідаційну групу і валідаційний комітет з організації В. Валідаційна група відповідає за затвердження (чи незатвердження) протоколів В.а.м. і дає остаточні рекомендації щодо результату В., планів її проведення і затвердження звітів і рекомендацій валідаційного комітету. До валідаційного комітету повинен входити співробітник, відповідальний за проведення В., та відповідальна особа з лабораторії контролю якості, яка має гарантувати, що методи випробувань, які використовуються для контролю якості, валідовані. Валідаційний комітет зобов’язаний звітувати перед валідаційною групою з виконання запланованих робіт. В.а.м. — обов’язковий елемент програми забезпечення якості, у зв’язку з чим вся документація з В. узгоджується і затверджується службою якості.
Гризодуб А.И. Валидация спектрофотометрических методик количественного анализа лекарственных средств в соответствии с требованиями ГФУ // Фармаком. — 2002. — № 3; Гризодуб А.И., Левин М.Г., Леонтьев Д.А. и др. Аттестация стандартных образцов. Сообщ. 1. Аттестация вторичных стандартных образцов для количественного хроматографического анализа лекарственных средств // Фармаком. — 1999. — № 2; Гризодуб А.И., Леонтьев Д.А., Доценко Т.Н., Денисенко Н.В. Критерии для параметров линейной зависимости при проведении валидации аналитических методик по ГФУ // Актуальні питання фармацевтичної і медичної науки і практики: Зб. наук. ст. — Запоріжжя, 2003. — Вип. Х; ДФУ. — Х., 2004; Зволінська Н.М., Архіпова Н.М., Леонтьєв Д.А., Гризодуб О.І. Створення Національної системи професійного тестування лабораторій контролю якості лікарських засобів: атестація зразків для кількісного спектрофотометричного аналізу // Фармац. журн. — 2003. — № 6; Метрологические аспекты официальных методик контроля качества лекарственных средств. 1. Методики ВЭЖХ // Фізіологічно активні речовини. — 2001. — № 1 (31); Daas A.G.J., Miller McB J.H. Relationship Between Content Limits, System Suitability for Precision and Acceptance / Rejection Criteria for Assays Using Chromatographic Methods // Pharmeuropa, December. — 1999. — Vol. 11, № 4; Supplementary Guidelines on Good Manufacturing Practice (GMP): Validation. — World Health Organization. QS/03.055/Rev. 1. — 2003.