
ЖИТТЄВИЙ ЦИКЛ ФАРМАЦЕВТИЧНОЇ ПРОДУКЦІЇ (ЖЦФП). Модель ЖЦФП побудована на базі аналізу основних стадій формування та зміни показників якості продукції й складається з ланцюжка послідовних видів діяльності, які впливають на якісні показники продукції. Відповідно до вимог міжнародних стандартів ISO серії 9000 «життєвий цикл» продукції (в ISO 9004 називається «петлею якості») складається із внутрішнього та зовнішнього кола. Внутрішнє коло петлі якості складається з таких етапів: планування → виробництво → експлуатація. Якість фармацевтичної продукції планується та формується у виробничій сфері й піддається змінам у споживчій сфері (зовнішнє коло) (рис. 1). Виробнича сфера: маркетинг та вивчення ринку, планування й розробка продукції, проектування й розробка процесів, матеріально-технічне забезпечення (закупівля матеріалів), виробництво, контроль, упакування, маркування, зберігання, збут, продаж. Споживча сфера: реалізація (обіг), монтаж й експлуатація обладнання, його технічне обслуговування, утилізація або вторинна переробка після завершення терміну служби.
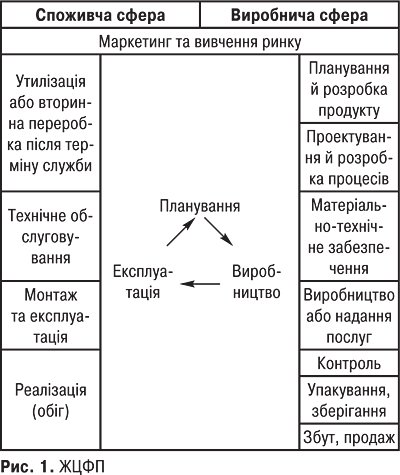
Забезпечення якості продукції здійснюється на всіх стадіях ЖЦФП, і всі види діяльності (наукові розробки — виробництво — реалізація — сервіс) впливають на якість кінцевого продукту. Для досягнення необхідної якості кожний етап бере участь у забезпеченні подальшого.
Вимоги до процесів, що формують якість на стадіях ЖЦФП
Планування якості (проектна якість) починається у процесі маркетингових досліджень — це перший фактор, що визначає якість майбутньої продукції (проводиться вивчення ринку, вирішується загальний задум товару, формується його образ і встановлюються загальні характеристики). Помилки на цьому етапі можуть бути найбільшими, оскільки якщо неправильно визначені потреби, наприкінці виробничого ланцюга можна одержати товар, який не користується попитом. На стадії планування проводиться комплекс заходів, спрямованих на запобігання помилкам при проектуванні, а також випробування і вимірювання параметрів продукції на різних етапах проектування. Виготовляється діюча модель або експериментальний зразок об’єкта в натуральну величину з реальними функціональними характеристиками та аналізується якість цього зразка, який дає можливість оцінити конструктивні рішення та якість вибраних матеріалів. Плануються всі процеси виготовлення та засоби контролю, які мають давати впевненість у тому, що технологічний процес і стан усіх елементів виробництва (устаткування, оснащення, інструменти і т.д.) забезпечать виготовлення фармацевтичного продукту (ФП) відповідно до вимог технічної документації.
Виробництво ФП — формування якості (виробнича якість) починається з моменту закупівлі матеріалів і закінчується моментом здачі продукції споживачеві (залежить не тільки від виробничих характеристик показників продукції, але й від якості пакування, якості збуту). Здійснюється випуск виробів запланованої якості із запланованою собівартістю. Контроль, проведення випробувань та досліджень — обстеження товару в процесі виробництва і вихідний контроль, що базуються на розвиненій метрологічній службі й орієнтовані на міжнародну сертифікацію якісної готової продукції та організаційно-технологічне забезпечення її виробництва. Стадія упакування та зберігання — це вирішення проблем збереження якості в період транспортування й зберігання товарів на основних і проміжних складах.
Реалізація (обіг) ФП починається від її відвантаження підприємством-виготовлювачем до одержання споживачем. Реалізація — це обмін і розподіл у разі незалежної системи збуту або розподіл і обмін при прямому маркетингу (за наявності у товаровиробників власної збутової мережі, напр. при фірмовій торгівлі). На стадії монтаж, експлуатація та технічне обслуговування ФП відбувається реалізація, підтримується й відновлюється їх якість (техобслуговування, ремонт, поставка запасних частин) протягом гарантійного терміну, що відображається в інструкціях, пам’ятках щодо догляду за виробом, настановах з експлуатації. Останньою стадією петлі якості продукції є утилізація або вторинна переробка. Якість цих процесів регламентується екологічними, санітарними й іншими нормами. Петля якості наочно показує послідовне відображення якості процесів на якості кінцевого результату. Неякісне виконання будь-якої з цих стадій не дозволяє досягти бажаної якості.
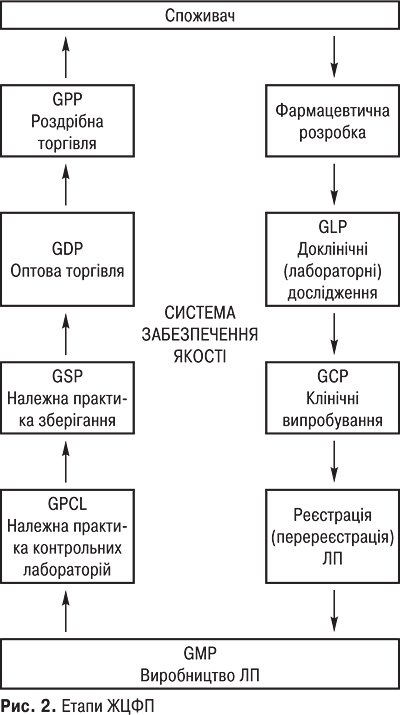
Сфера ЖЦФП має такі стадії: фармацевтична розробка, доклінічні дослідження, клінічні випробування, виробництво, дистрибуція, зберігання, увезення в Україну, вивезення із України, реалізація, застосування, утилізація ЛП. Вимоги кожного етапу ЖЦФП (петлі якості) відображені у відповідних Належних практиках (GXP). А саме: Настанова з якості, Фармацевтична розробка, Належна лабораторна практика (GLP), Належна клінічна практика (GCP), Реєстрація або ліцензування (перереєстрація) ЛП (Good Practice Registration — GPR), Належна виробнича практика (GMP), Належна практика контрольних лабораторій (Good Practices for National Drug Control Laboratories — GPCL), Належна практика зберігання фармацевтичної продукції (Guide to good storage practices for pharmaceuticals — GSP), Належна практика дистрибуції (GDP), Належна аптечна практика (GPP) (рис. 2).
Необхідні споживчі властивості ЛП (якість, ефективність, безпека) формуються в ході їхньої розробки й випробувань, з дотриманням правил GLP і GСР, фіксуються актом їхньої реєстрації. У процесі серійного виробництва ці властивості формуються за допомогою дотримання правил GMP, а також державного інспектування й зберігаються в мережі розподілу завдяки впровадженню правил GDP й GPP. На кожному етапі створення, виготовлення й розподілу ЛП здійснюються специфічні заходи недопущення помилок і відхилень у роботі, які можуть негативно вплинути на якість, враховуються помилки, що також впливають на якість ЛП (приміщення, персонал, устаткування, організація та ведення технологічного процесу, документація, контроль процесу виробництва, контроль якості готового продукту тощо). Правила GMP вимагають, щоб нові препарати, передані в серійне виробництво, були розроблені й випробувані відповідно до правил GLP й GCP. У свою чергу, передбачено, що біохімічні й інші лабораторії, які беруть участь у доклінічних випробуваннях препаратів, відповідали б вимогам GLP. Аналогічні вимоги все більше поширюються й на аналітичні лабораторії, які здійснюють контроль якості ліків. Крім того, продукція, виготовлена відповідно до правил GMP, може зіпсуватися протягом дистрибуції, якщо не будуть дотримані відповідні вимоги, стандарти GDP. Відображаючи різні аспекти єдиної концепції забезпечення якості, ефективності й безпеки ліків, правила GХР тісно пов’язані між собою внутрішньою логікою й підходами та засновані на комплексному обліку всіх факторів, здатних негативно вплинути на якість ЛП. Таким чином, можна говорити про ланцюжок забезпечення якості, що охоплює весь ЖЦФП, суттю якого є послідовність і безперервність.
Левашова И.Г., Мурашко А.Н., Подпружников Ю.В. Надлежащие практики в фармации. — К., 2006.