
ЗАГАЛЬНИЙ ТЕХНІЧНИЙ ДОКУМЕНТ (Common Technical Document — CTD) — нормативний документ-настанова, що регламентує загальну форму складання заявки на отримання торгової ліцензії на ЛП (структуру реєстраційного досьє) незалежно від процедури (централізованої, взаємного визнання або національної) і від обсягу заявки (повний або скорочений варіант), а також дає рекомендації, якими нормативними документами (настановами з якості, біотехнології, фармакології, токсикології та ін.) слід користуватися при проведенні досліджень і складанні розділів реєстраційного досьє. З.т.д. був розроблений в рамках процесу ICH (The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use) та опублікований у 2002 р. в томі 2В (Інформація для заявників: надання та зміст реєстраційного досьє) Правил, регулюючих ЛП в ЄС. Він призначений для економії часу і ресурсів, а також для полегшення процесів рецензування та обміну інформацією. Форма представлення документації у вигляді версії З.т.д.-формату застосовується для підготовки добре структурованих заявок до всіх типів препаратів (нові хімічні речовини, радіофармацевтичні препарати, вакцини, фітопрепарати та ін.), які подаються компетентним уповноваженим органам на отримання торгової ліцензії в 3 регіонах ICH — Європі, США та Японії. З.т.д. складається з 5 модулів (рисунок). Зміст модуля 1 визначений Європейською комісією при консультативній допомозі з боку компетентних уповноважених органів держав — членів ЄС, Європейського агентства з оцінки ЛП та ін. зацікавлених сторін. Модулі 2, 3, 4, 5 є однаковими для всіх регіонів. Адміністративна, регіональна і національна інформація міститься в модулі 1 (форма заявки на отримання торгової ліцензії, коротка характеристика ЛП, запропонована заявником, маркування, анотація-вкладиш в упаковку та ін.). До модулю 2 входять: загальне резюме з якості, огляд/резюме доклінічних даних та огляд/резюме клінічних даних, які повинні бути підготовлені кваліфікованими та досвідченими експертами. Експерти повинні поставити підпис і навести коротку інформацію в спеціальному розділі модуля 1 про рівень своєї професійної освіти і спеціальних знань. У модулі 3 наводять документацію з хімічних, фармацевтичних та біологічних властивостей препарату. Ця інформація повинна бути структурована відповідно до вимог Настанови M4Q ICH Harmonised Tripartite Guideline, Common Technical Document, Module 2: Quality Overall Summary (QOS), Module 3: Quality, 2000. Документація, що стосується токсикологічних та фармакологічних досліджень, виконаних на активній субстанції та готовій лікарській формі препарату, повинна бути наведена в резюме доклінічних даних у текстовому форматі (модуль 2) та у звітах про доклінічні дослідження (модуль 4). Ці звіти повинні відповідати Настанові M4S ICH Harmonised Tripartite Guideline, Common Technical Document for Registration Pharmaceuticals for Human Use, Safety: Nonclinical Summaries and Organisation Module 4, 2000. Документація з клінічних випробувань, що проводились з використанням готової лікарської форми препарату, повинна бути надана в резюме клінічних даних у текстовому форматі (модуль 2) та у звітах про клінічні дослідження (модуль 5). Ці звіти повинні відповідати Настанові М4Е ICH Harmonised Tripartite Guideline, Common Technical Document for Registration Pharmaceuticals for Human Use, Efficacy: Module 2: Clinical Overview and Clinical Summary, Module 5: Clinical Study Reports, 2000.
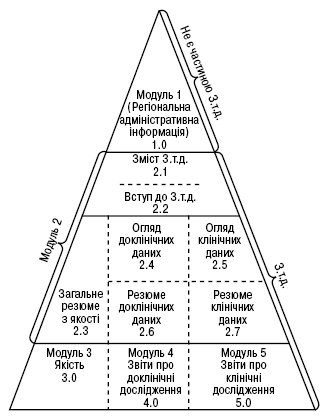
Рисунок. Структура З.т.д.
Міжнародний формат З.т.д. призначений для всіх категорій ЛП і всіх типів заявок на отримання торгової ліцензії. Якщо у відповідному розділі заявки відсутня інформація або в ній немає необхідності, то такий розділ позначають як «не застосовується» або не «відповідає», при цьому зберігається назва розділу та нумерація. Відсутність досліджень слід обгрунтувати у відповідному розділі загального резюме з якості, огляду доклінічних або клінічних даних. Для бібліографічних та скорочених заявок необхідно мати згоду власника торгової ліцензії та підстави для заяви про те, що ЛП є по суті аналогічним. Обов’язок заявника на отримання торгової ліцензії на ЛП — гарантувати надання компетентним уповноваженим органам повної інформації про препарат. Дані, що стосуються процесу виробництва АФІ, дозволяється подавати в окремій конфіденційній документації. Отже, заявник при наданні окремого майстер-файла на ЛП (Drug Master File — DMF) у форматі З.т.д. повинен консультуватися та працювати з відповідальною особою виробника активного інгредієнта для гарантії того, що вся необхідна інформація надається безпосередньо до компетентних уповноважених органів. Даний DMF повинен включати відповідні частини модуля 3, тобто детальний опис процесу виробництва, контролю якості під час виробництва, процесу валідації та оцінки даних. Крім того, необхідно надати окремо загальне резюме з якості (модуль 2), в якому навести всі ті аспекти, які не увійшли до заявки на отримання торгової ліцензії на ЛП. DMF повинен повністю відповідати З.т.д.-формату. При складанні досьє для подання заявки на отримання торгової ліцензії заявники повинні враховувати вимоги настанови, відносно якості, безпеки й ефективності ЛП, опублікованих комісією в «Правилах, регулюючих лікарські препарати в Європейському співтоваристві» (томи ЗА, ЗВ, ЗС): Настанови з якості, безпеки та ефективності ЛП для людини прийняті Комітетом із патентованих ЛП (Committee for Proprietary Medicinal Products — CPMP). При складанні частини досьє, що належить до якості, також можна використовувати монографії й загальні розділи Європейської Фармакопеї (Європейський сертифікат відповідності монографії). Усі речовини, одержані від жуйних тварин, додатково повинні відповідати вимогам безпеки відносно ризику передачі трансмісивних губчастих енцефалопатій (Transmissible Spongiform Encephalopathies — TSE). Представлення інформації в документації З.т.д.-формату повинно бути точним і ясним для полегшення перевірки основних даних і для того, щоб рецензент міг швидко орієнтуватися у змісті заявки. При складанні реєстраційного досьє для ліцензування препарату в конкретному регіоні необхідно внести зміни тільки у «верхівку айсберга» — модуль 1 (регіональна адміністративна інформація). Форма і структура реєстраційного досьє у форматі З.т.д. значно зменшує час та ресурси, що використовуються для складання заявок на реєстрацію ЛП для людини. Завдяки введенню З.т.д. будуть полегшені як процес експертизи в регуляторних органах, так і спілкування з заявниками. Крім того, спрощений обмін інформацією між регуляторними уповноваженими органами.
Структура реєстраційного досьє, прийнята в Україні, дозволяє використання формату З.т.д. за бажанням заявника, але нині він не відповідає в повному обсязі З.т.д. як за змістом, так і за формою.
Фармацевтический сектор: общий технический документ для лицензирования лекарственных средств в ЕС / Авт.-сост.: В.А. Усенко, Н.А. Ляпунов, Е.П. Безуглая, А.Л. Спасокукоцкий, Т.К. Ефимцева; под редакцией: А.В. Стефанова, Т.А. Бухтиаровой, В.Г. Варченко и др. — К., 2002; Rules Governing Medicinal Products in European Union, vol. 2A, 2002.